








Software/Method Development
- is a versatile and widely used molecular simulation program with broad application to many-particle systems
- has been developed with a primary focus on the study of molecules of biological interest, including peptides, proteins, prosthetic groups, small molecule ligands, nucleic acids, lipids, and carbohydrates, as they occur in solution, crystals, and membrane environments
- provides a large suite of computational tools that encompass numerous conformational and path sampling methods, free energy estimates, molecular minimization, dynamics, and analysis techniques, and model-building capabilities
- is useful for a much broader class of many-particle systems
- can be utilized with various energy functions and models, from mixed quantum mechanical-molecular mechanical force fields, to all-atom classical potentials with explicit solvent and various boundary conditions, to implicit solvent and membrane models
- has been ported to numerous platforms in both serial and parallel architectures
B. R. Brooks, C. L. Brooks III, A. D. MacKerell, Jr., L. Nilsson, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, L. Caves, Q. Cui, A. Dinner, S.
Fischer, J. Gao, M. Hodoscek, K. Kuczera, T. Lazaridis, J. Ma, E. Paci, R. W. Pastor, C. B. Post, M. Schaefer, B. Tidor, R. M. Venable, H. L.
Woodcock, X. Wu and M. Karplus; CHARMM The Biomolecular Simulation Program. J. Comp. Chem.; 2009; 30 (10); 1545-1614.
Q-Chem is a comprehensive ab initio quantum chemistry package. Its capabilities range from the highest performance DFT/HF calculations to high level post-HF correlation methods. Q-Chem tackles a wide range of problems in commercial, academic and government laboratories, including:
|
|

Y. Shao, L. Fusti-Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. lipchenko, S. V. Levchenko, D. P. ONeill, R. A.
Distasio, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P.
E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Daschel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani,
S. R. Gwaltney, A. Heyden, S. Hirata, C. P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E.
I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, D. C. Sherrill, A. C. Simmonett, J. E. Subotnik, H. Lee Woodcock, W. Zhang, A. T.
Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer, J. Kong, A. I. Krylov, P. M. W. Gill, M. Head-Gordon; Advances in
methods and algorithms in a modern quantum chemistry program package. (Phys. Chem. Chem. Phys.); 2006; 8; 3172-3191.
A new web portal for the CHARMM macromolecular modeling package, CHARMMing (CHARMM interface and graphics, http://www.charmming.org), is presented. This tool provides a user friendly interface for the preparation, submission, monitoring, and visualization of molecular simulations (i.e., energy minimization, solvation, and dynamics). The infrastructure used to implement the web application is described. Two additional programs have been developed and integrated with CHARMMing: GENRTF, which is employed to define structural features not supported by the standard CHARMM force field, and a job broker, which is used to provide a portable method for using grid and cluster computing with CHARMMing. The use of the program is described with three proteins: 1YJP, 1O1O and 1UFY. Source code is provided allowing CHARMMing to be downloaded, installed, and used by supercomputing centers and research groups that have a CHARMM license. Although no software can replace a scientist's own judgment and experience, CHARMMing eases the introduction of newcomers to the molecular modeling discipline by providing a graphical method for running simulations.
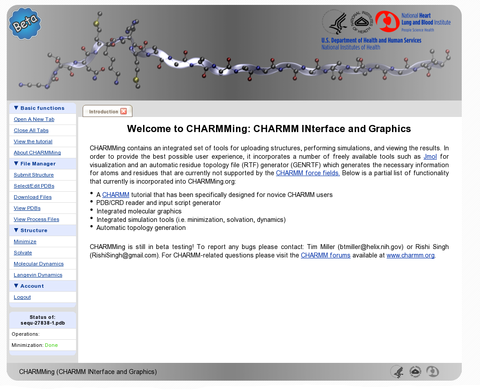
B. T. Miller, R. P. Singh, J. B. Klauda, M. Hodoscek, B. R. Brooks, and H. Lee Woodcock* CHARMMing: A New, Flexible, Web-based front-end to
CHARMM. J. Chem. Inf. Mod.; 2008; 48(9); 1920-1929.
CHARMM Tutorial
Rishi P. Singh, Benjamin T. Miller, Jeffery Klauda, Xiongwu Wu, and H. Lee Woodcock
A tutorial/introduction to the use of CHARMM has been created and published on the web (CHARMM Tutorial). In addition, this has been integrated into CHARMMing. This tutorial provides conceptual foundations to molecular simulation techniques such as energy minimization, solvation, molecular dynamics, and the use of periodic boundary conditions that are implemented in CHARMMing. Thus CHARMMing may be used as an introduction to molecular simulations in general and CHARMM in particular with the tutorial serving as a manual.
